
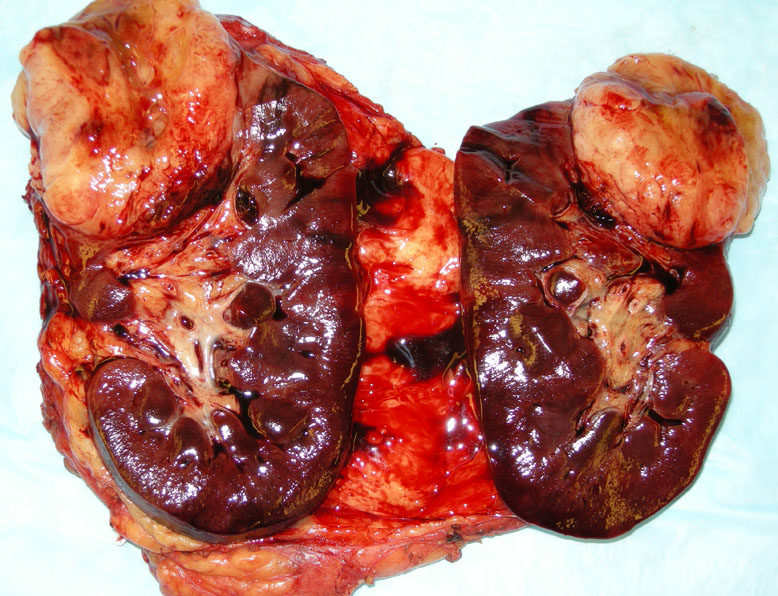
The upper pole of the kidney contains a bulging firm mass with a mesenchymal appearance.
){kind=link}
This synovial sarcoma (SS) was from an 18 year old male, and the tumor did demonstrate t(x;18) by FISH. Normal kidney (lower half) interfaces with the sarcoma, which shows a pushing border.
){kind=link}
This monophasic SS exhibits spindled cells arranged in short fascicles. The background fibrovascular stroma contains narrow, tapered fibroblasts and small vessels. Although not seen in this image, a branching pericytoma-like vascular pattern is often seen associated with this tumor. Mitotic figures can be variable. Note the mitotic figure in the lower right.
){kind=link}
A closer view demonstrates the spindled cells, which are rounded, but can be distinguished from epithelial cells by their scant cytoplasm and stippled chromatin pattern. Occasional mast cells can be seen in the stroma.
){kind=link}
Synovial sarcoma (SS), the fourth most common sarcoma (~10-15% of sarcomas), was originally thought to be derived from synovial cells due to their propensity to be located next to joints; furthermore, an epithelial component was often seen on light microscopy. However, this theory has been disproved as the epithelial component of SS express certain antigens (EMA, cytokeratins) not found in synovial cells.1 It is no longer believed that SS is related to synovium or percursor synoviocytes, however, the cell of origin is still under investigation.
There are two main variants of SS: biphasic and monophasic. Both forms contain the spindled component composed of plump to tapered spindled cells arranged in short fascicles. The cells have dark nuclei with a stippled chromatin pattern with scant cytoplasm and indistinct cell borders. The biphasic form contains an added population of epithelial cells consisting of cuboidal or columnar cells forming glandular structures or less commonly, solid nests. The epithelial cells are recognized by their larger and paler morphology; the nuclei are round and vesicular, the cell borders are more distinct and they have more cytoplasm.
The cytogenetic hallmark for SS is the t(X;18)(p11.2;q11.2) translocation. The SSX1 or SSX2 gene located on chromosome Xp11 is fused to the SYT gene on chromosome 18q11. This fusion gene product encodes a chimeric protein that activates transcription. In a study of 22 biphasic and 64 monophasic SS, Guillou et al (2001) reports a sensitivity and specificity of 100% and 96% using RT-PCR for t(X;18).3
The differential diagnosis for the monophasic spindled SS includes malignant peripheral nerve sheath tumor (MPNST) or fibrosarcoma. IHC stains will be helpful in sorting out these entities, as SS will demonstrate at least focal positivity for EMA and keratin (which are generally negative in MPNST and fibrosarcoma).2,4
A relatively new immunostain that may be helpful in the diagnosis of SS is TLE1. Most other spindle cell tumors on the differential will be negative except for a subset of hemangiopericytomas, solitary fibrous tumors and schwannomas (Miller).
This tumor chiefly occurs in teenagers and young adults with a slight male predilection (peak incidence between 10-35).2 The majority of tumors (60-70%) develop in the lower extremity with the knee and thigh being the most common sites. Note, however, that SS is a ubiquitous tumor and can arise anywhere in the body such as trunk, head and neck and viscera (ie. lung/pleura, kidney GI tract). Patients often have a history of a deep-seated mass that has been present for years.
Surgery and adjuvant chemotherapy.
5 year survival rate is 60-65%, but 10 year survival drops to 30%.2 Favorable prognostic factors include smaller tumor size (less than 5 cm), younger age at diagnosis (less than 10 years old), earlier clinical stage at presentation and lower histological grade as defined by presence of mitosis and necrosis.2 Some have reported that patients with the SYT-SSX2 fusion gene have a more favorable outcome than patients with the SYT-SS1 fusion gene, however, this remains an area of dispute.1,2
→The cytogenetic hallmark for SS is the t(X;18)(p11.2;q11.2) translocation
→TLE1 is a key immunostain for SS.
• Uncertain Lineage : Synovial Sarcoma
1 Kempson RL, Fletcher CDM, Evans HL, Hendrickson MR, Sibley RK. Tumors of the Soft Tissues: Atlas of Tumor Pathology. Third Series, Fascicle 30. Washington DC: AFIP; 1998: 472-484.
2 Fletcher CDM, ed. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Elsevier; 2007: 1576-8.
3 Guillou L et al. Detection of the Synovial Sarcoma Translocation t(X;19)(SYT;SSX) in Paraffin-Embedded Tissues Using Reverse Transcriptase-Polymerase Chain Reaction: A Reliable and Powerful Diagnostic Tool for Pathologsts. Hum Pathol (2001) 32:105-112.
4 Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005: 1323.
Miller RT. TLE1 Immunostains in the Diagnosis of Synovial Sarcoma. May 2007: ProPath Immunohistochemistry. Available at www.ihcworld.com/_newsletter/2007/2007_05_TLE1_synovial_sarcoma_lowres.pdf