
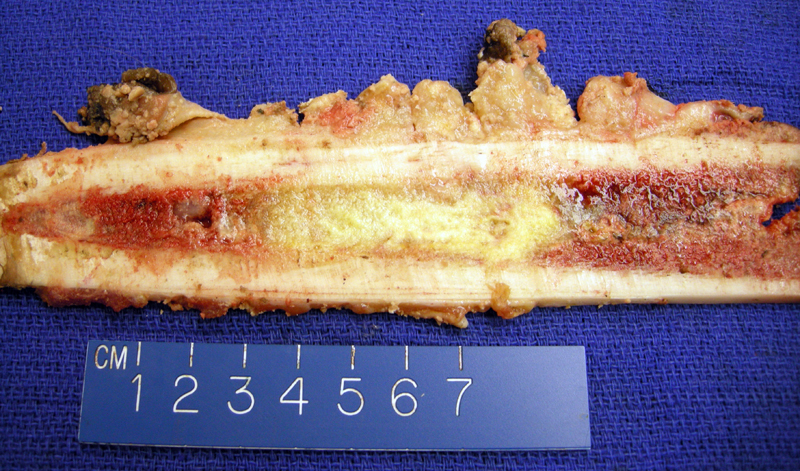
The femur is involved by an intramedullar mass which also extended into the soft tissue.
Often, the gross specimen obtained in a Ewing sarcoma is post chemotherapy, as the usual treatment protocol consists of (1) induction chemotherapy to achieve regression and local control of the tumor; (2) resection of tumor; (3) continuation of chemotherapy. This specimen was post-chemotherapy and thus, largely necrotic.
A touch prep demonstrates the cytologic features of ESFT, with cohesive cluster of cells containing scant cytoplasm, indistinct nucleoli and a finely granular chromatin pattern.
One might even image a rosette formation in this touch prep.
Microscopically, Ewing sarcoma is composed of small round blue cells that are slightly larger than lymphocytes. A lobular or trabecular arrangement is most common, however, this image demonstrates more of a diffuse pattern of growth.
The malignant cells have pale, uniform round nuclei with a finely granular chromatin pattern.
Tumor cells surrounding bone is seen. Although some of the cells may look slightly spindled (probably due to artifactual crushing), a spindled morphology is not part of ESFT and if definite spindling is seen, another diagnosis must be considered.
This image demonstrates malignant cells with more abundant cytoplasm and more distinct cell borders. One might argue that this portion of the tumor exhibits more epithelial differentiation consistent of the the historical definition of PNET.
The cells in this femoral Ewing's sarcoma are compartmentalized by a fibrous stroma. The cells themselves appear relatively uniform and contain a large round nucleus occupying most of the cell.
CD99 shows strong membranous staining of tumor cells.
FISH shows the EWS breakpoint on chromosome 22. When the two colors (red and green) are superimposed on other another, it creates a yellow spot, signifying an intact EWS locus. However, when they break apart (red and green are separated), it demonstrates that the EWS gene has been rearranged and translocation has occured. The most common tranlocation involves the EWS gene (22q12) to the FLI1 gene (11q24). This t(11;22)(q24;q12) accounts for 90-95% of ESFT. In 5-10% of cases, the translocation is t(21;22)(q21;12). Less than 1% of tumors demonstrate a t(7;22)(q22;12) translocation.
Karyotype demonstrating the t(11;22)(q24;q12) translocation.
Ewing sarcoma family of tumors (ESFT) encompasses the following entities: Ewing sarcoma, primitive neuroectodermal tumor (PNET) and malignant small cell tumor of the thoracopulmonary region (Askin tumor). ESFT all share the common molecular alteration of the translocation of the EWS gene (located on 22q12) to a transcription factor belonging to the ETS family (mainly FLT1). The most common translocation (90-95% of ESFT) is t(11;22)(q24;q12), creating the fusion gene EWS-FLT1 that acts as an oncogene and stimulates cell proliferation. The second most common translocation (~5% of ESFT) is t(21;22)(q22;q12), where EWS on 22q12 is translocated to the ERG at 21q22 to create the fusion gene EWS-ERG.
Historically, it was thought that tumors in the bone (Ewing sarcoma) were largely undifferentiated, and peripheral or primitive neuroectodermal tumors (PNET) arising in the soft tissues demonstrated neuroepithelial differentation. Microscopic neural features would include Homer-Wright rosettes and pseudorosette formation -- epithelial features would include prominent nuclear membranes, abundant cytoplasm and glandular formation. However, considerable overlap exists and it is not uncommon to see rosettes in Ewing sarcoma or an undifferentiated morphology in PNET. Therefore, many authorities simply refer to the tumors as Ewing Sarcoma Family of Tumors or Ewing Sarcoma/PNET, acknowledging that while Ewing Sarcoma is more poorly differentiated than PNET, the features often meld together and strict distinctions cannot be drawn.1,4
As with all small round blue cell tumors, the list of differential diagnoses is long (neuroblastoma, lymphoma, small cell carcinoma, rhabdomyosarcoma, etc...), therefore, aside from molecular studies, one will have to perform a panel of IHC stains. The hallmark of ESFT is positivity for CD99 (which is encoded by the MIC2 gene located in the short arms of the X and Y chromosome). 90% of ESFT demonstrates membranous staining for CD99. It is important to note, however, that CD99 is sensitive, but not specific for ESFT, as many other tumors such as rhabdomyosarcoma, neuroblastoma, lymphoblastic lymphoma may also be positive for CD99. Fortunately, most of these other tumors will also demonstrate positivity in antigens that ESFT will be negative for (ie. neuroblastoma is positive for synaptophysin, but ESFT is not).
80% of affected patients are younger than 20 years old, and most of the affected patients are between 10-15 years old. Boys are more commonly affected than girls, with a reported ratio of ~1.5:1.1 The disease preferentially affects whites, as the disease is exeedingly rare in blacks.
Presents as painful, warm swollen masses in affected sites (most commonly diaphysis of long bones such as the femur), and may be accompanied by fever, elevated sedimentation rate and leukocytosis. These constellation of symptoms may often be misdiagnosed initially as osteomyelitis.
Note that although ESFT is classically described as involving bone, extraosseous ESFT are not uncommon and can arise anywhere in the body. Cases of extraosseous ESFT have been described involving the deep soft tissue, skin, visceral organs and arising from a nerve. ESFT located in the chest wall involving chest, pleura and lungs are called "Askin tumors".3
Radiographs reveal a destructive lesion that permeates through the cortical bone into the surrounding soft tissue. There may be a reactive periosteal bone formation; layers of new bone is added to create an "onion-skin" appearance.
Before the advent of radiation and chemotherapy, the 5 year survival rate was 5-10%, and now, it has improved to 50-60%.3 Prognosis is highly dependent on whether the disease is localized or metastatic at the time of presentation. In a study of 181 patients with ESFT, the 5 and 10 year survival rates for those with localized disease was 73% and 65% respectively, compared to 35% and 23% for those with metastatic disease.1
Tumor response to chemotherapy (measured by the percentage of necrosis in the tumor) is also considered a prognostic factor.
• Chondroid : Chondrosarcoma, Mesenchymal Type
1 Khoury JD. Ewing Sarcoma Family of Tumors. Adv Anat Pathol (2005)12:212-220.
2 Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005: 1301-2.
3 Fletcher CDM, ed. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Elsevier; 2007: 1600-2,1750-1753.
4 Rosai, J. Rosai and Ackerman's Surgical Pathology. 9th Ed. Philadelphia, PA: Elsevier; 2004: 2172-76.
***FISH and karyotype images courtesy of Skip Haines, Tricore Reference Laboratories, Albuquerque NM.
***Gross images courtesy of Myra Zucker, Dept of Pathology at University of New Mexico, Albuquerque NM.

){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
 arrows.JPG', 'Translocation between chromosomes 11 and 22.', 'cytogenetics', '')){kind=link}