
Author: Cecilia Wu, MD, Dept of Pathology, University of New Mexico
Co-author(s): Lisa Cerilli, MD, Dept of Pathology, University of New Mexico
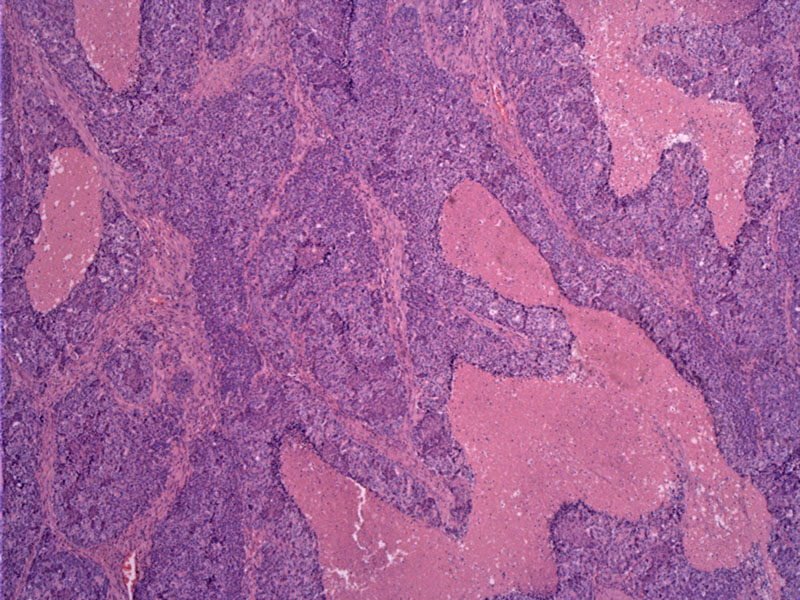
Uniform undifferentiated epithelial cells grow in undulating sheets (purple confluent regions) with areas of geographic tumor necrosis (pink).
In this image, small blue primitive cells are closely packed, and grow in broad trabeculae.
Nests of cells differentiate towards acinar structures, with formation of small lumens. The tumor may be AFP positive.
Well-defined acinar structure can be discerned. Intimately admixed squamoid areas (cells with more abundant pink cytoplasm) are also noted, another feature of this tumor. Although not obvious in this image, squamoid morules can be especially helpful in distinguishing between PB and acinar cell carcinoma.
Pancreatoblastoma (PB) is an extremely rare pancreatic tumor of childhood. PB is associated with Beckwith-Wiedemann syndrome and familial adenomatous polyposis. Molecular aberrations discovered thus far include alterations in the Wnt signaling pathway as well as chromosome 11p loss of heterozygosity (Saif). The tumor is capable of showing multiple lines of differentiation including acinar, squamous and endocrine as well as ductal.
Grossly, PB are well-circumscribed, multi-lobulated and quite large (mean size of 10.5 cm). Histologically, sheets of uniform neoplastic cells demonstrate acinar (most pervasive) and squamoid differentiation. Sometimes, ductal, endocrine and mesenchymal differentation can be seen as well (Iacobuzio-Donahue). A key feature is the formation of squamous corpuscles.
Pancreatoblastoma mainly presents during childhood but approximately one third occur in adults, thus with a bimodal at age 4 and in the third decade. Children usually present with upper abdominal pain and a palpable mass. Tumors usually arise in the pancreatic head, in which case they may cause obstruction of the upper duodenum and gastric outlet resulting in vomiting, jaundice and gastrointestinal bleeding. Serum alpha-fetoprotein can be elevated in up to 68% of patients (Saif).
Treatment is complete resection, which is often curative. The role of adjuvant chemotherapy or radiotherapy is uncertain due to the small number of patients treated. Chemotherapy regimens consisting of cyclophosphamide, etoposide, doxorubicin, and cisplatin have been used in neoadjuvant setting with anecdotal benefit. Prognosis of this rare tumor is good, when resected completely.
PB tend to be less aggressive in infants and children compared to adults. In adults, median survival time of 18.5 months has been reported (Cavallini). Prognosis overall is quite favorable, when resected completely. Prognosis is poorer, when there is metastasis or when it is inoperable.
→PB is a tumor of childhood, but can occur in up to 1/3 of adults.
→Usually presents with a palpable mass in epigastrium in a child. Some have elevated AFP levels.
→PB is less aggressive in children than in adults.
→Histologically, nests of tumor cells form islands and show acinar and squamoid differentiation.
→Squamoid nests help distinguish between acinar cell carcinoma and acinar cell carcinoma.
• Pancreas : Acinar Cell Carcinoma
Cavallini A, et al. Pancreatoblastoma in adults: a review of the literature. Pancreatology. 2009;9(1-2):73-80.
Iacobuzio-Donahue CA, Montgomery EA. Gastrointestinal and Liver Pathology: Foundations in Diagnostic Pathology. Philadelphia, PA: Elsevier; 2005: 479.
Saif MW. Pancreatoblastoma. JOP. 2007 Jan 9;8(1):55-63.

){kind=link}
){kind=link}
){kind=link}
){kind=link}