
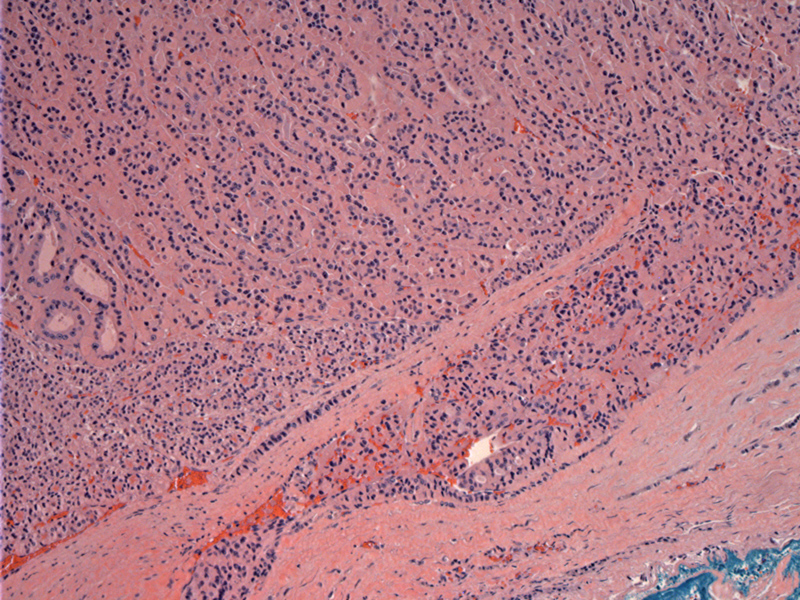
The tumor has permeated into its thick capsule, with an area of lymphatic involvement (bottom image). Criteria for malignancy includes one or more of the following : capsular invasion that penetrates the full thickness of the capsule; spread to adjacent tissue; vascular invasion; and local or distant metastases.
){kind=link}
Other areas are more convincing for vascular invasion.
){kind=link}
The tumor itself consists of sheets of oncocytic cells, with some follicular development.
){kind=link}
Despite the World Health Organization's inclusion of this entity as follicular cell cancer (FCC), some authors assert that Hurthle cell carcinoma (HCC) remains a distinct clinical entity. For example, only ~10% of metastases from HCC take up radioiodine, compared with 75% of metastases from FCC (Yutan). Compared to FCC, HCC is more often multifocal, bilateral and more frequently involves regional nodes. Thus, HCC appears to be a more aggressive tumor than FCC, with a greater tendency to metastasize to distant sites and a higher mortality rate (Yutan).
The key feature distinguishing Hurthle cell carcinoma from Hurthle cell adenoma is the presence of capsular or vascular invasion. Furthermore, Hurthle cell carcinoma tend to be larger and the age at presentation tends to be older compared to Hurthle cell adenoma(Zhang, DeLellis). A size greater than 4 cm is strongly correlated with malignancy (Fletcher).
Grossly, these tumors are brown due to their abundant mitochondria. Histologically, greater than 75% of the tumor should be composed of oncocytic cells with deeply eosinophilic granular cytoplasm and round nuclei containing a centrally placed nucleoli (Thompson). Occasionally, the cytoplasm may be clear and vacuolated due to ballooning of the mitochondria. The oncocytic cells form follicles, trabeculae or solid sheets. The colloid may be calcified and mistaken for psamomma bodies (Fletcher).
The peak age of onset for HCC is about a decade later than conventional follicular carcinomas. In one cohort, the peak age of diagnosis of HCC was 59 compared to 35-45 for follicular carcinoma (Mills). Hürthle cell carcinoma is more likely to be familial than FCC. Up to 39% of patients in one study reported childhood head and neck radiation (Yutan).
Distant metastases are found in approximately 10% to 29% at initial diagnosis of HCC (Yutan).
Initial treatment of an FNA lesion regarded as a Hurthle cell neoplasm is lobectomy and isthmectomy. If there is a history of radiation to the head or neck or a family history of HCC, a total thyroidectomy should be performed.
Once a diagnosis of Hurthle cell carcinoma is established, complete thyroidectomy is recommended. The Delphian and ipsilateral perithyroidal and central neck nodes should be removed, as these tumors are usually unresponsive to radioiodine. Completeness of thyroidectomy should be evaluated by radioiodine scan 3 to 4 months after surgery (Yutan).
Thyrotropin-suppressive doses of thyroid hormone should be given because most of these tumors have thyroid stimulating hormone (TSH) receptors.
A recent review of 62 patients with HCC confirmed previous findings that it is a more aggressive variant. For example, 20% of the HCC cases demonstrated nodal metastases whereas nodal disease is rare in conventional follicular carcinoma. The recurrence rate at 30 years is double that of conventional follicular carcinoma (58% vs. 30%). The survival rate is also reduced in HCC comparatively (64% vs. 79%)(Mills).
In another review of 55 patients, older age and large tumor size were two factors that predicted malignancy (Zhang).
→Though often classified as a variant of follicular cell carcinoma, Hurthle cell carcinoma is generally more aggressive.
&rarr:As with follicular neoplasms, the presence of capsular or vascular invasion distinguishes Hurthle cell carcinoma from Hurthle cell adenoma.
→A size greater than 4 cm is suggestive of malignancy.
→Hurthle cell carcinomas are more likely to be multifocal and bilateral, especially in the setting of prior head and neck radiation.
• Thyroid : Hurthle Cell Adenoma
DeLellis RA, Lloyd RV, Heitz PU, Eng C. Tumors of Endocrine Organs: WHO Classification of Tumours. Lyon; IARC Press; 2004: 67-72.
Fletcher CDM, ed. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Elsevier; 2007: 1026-8.
Mills SC, Haq M, Smellie WJ, Harmer C. Hürthle cell carcinoma of the thyroid: Retrospective review of 62 patients treated at the Royal Marsden Hospital between 1946 and 2003. Eur J Surg Oncol. 2009 Mar;35(3):230-4. Epub 2008 Aug 21.
Thompson LDR. Endocrine Pathology: Foundations in Diagnostic Pathology. Philadelphia, PA: Elsevier; 2006: 51-64, 101-8.
Zhang YW, Greenblatt DY, Repplinger D et al. Older age and larger tumor size predict malignancy in hürthle cell neoplasms of the thyroid. Ann Surg Oncol. 2008 Oct;15(10):2842-6.
Yutan E, Clark OH. Hürthle cell carcinoma. Curr Treat Options Oncol. 2001 Aug;2(4):331-5.