
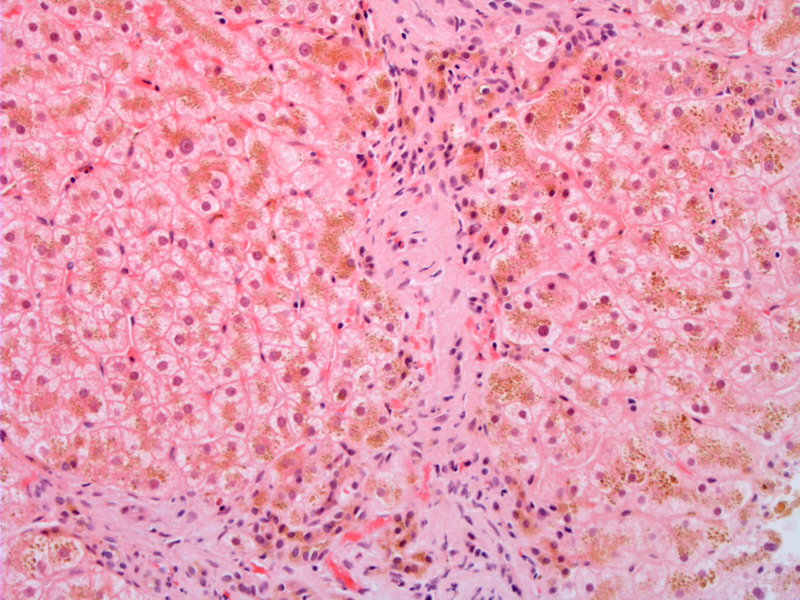
Heavy iron deposition is seen in hepatocytes as golden-yellow granules in the cytoplasm.
){kind=link}
Septal fibrosis (left) is indicative of early cirrhosis.
){kind=link}
Staining using Prussian blue demonstrates heavy iron deposition, especially in the periportal area.
){kind=link}
Using PCR, the area of interest (codon 282 and 63 of the short arm of chromosome 6) is amplified in two separate reactions. In this particular assay, a restriction enzyme will make a cut at the mutation site of 282 if the mutation is PRESENT and another restriction enzyme will make a cut at the mutation site of 63 if the mutation is NOT PRESENT. Confusing, I know, but just look at the band patterns and compare them to controls. Then, the DNA pieces are run on an agarose gel to separate the pieces according to size. So the lane for the C282 assay (on the left) labeled
){kind=link}
In this separate assay, lanes 4 and 5 are heterozygous for C282Y and no one has mutations for H63D.
){kind=link}
Hemochromatosis can be primary (inherited) or secondary (acquired). Inherited hemochromatosis is due to an autosomal recessive genetic disorder that leads to excessive iron absorption. The hemochromatosis gene (HFE) is located on chromosome 6, close to the HLA gene locus -- this gene is involved in the regulation of dietary iron absorption by intestinal epithelial cells.
The most common defect involves a cysteine-to-tyrosine substitution at amino acid 282, or C282Y. In persons of Northern European extraction, the prevalence of homozygotes is estimated to be 1 out of every 200 persons) and the prevalence of heterozygotes is approximately 1 out of every 10 persons -- thus, hereditary hemochromatosis is one of the most common genetic disorders in whites. Frequency and allele analysis suggest a Celtic origin, and there may have been a selection advantage for iron retention, hence decreased iron-deficiency.2
It is important to keep in mind, however, that the penetrance of the disease is only ~20% in homozygotes, therefore, only not everyone with the mutation manifests the disease.1 Other less common mutations include the gene H63D and S65C.
In normal adults, the total body iron ought to be 2-6 grams. In hereditary hemochromatosis, total iron may exceed 50 grams, leading to excess iron deposition in the liver, pancreas, endocrine organs (adrenals, thyroid and pituitary), heart and skin. Microscopically in the liver, iron deposits appear as golden granules in the cytoplasm in the periportal hepatocytes. As the disease progresses, the deposits appear in the rest of the lobule, including Kupffer cells and bile duct epithelium.1 Eventually, as excessive iron is hepatotoxic and stimulates the formation of collagen, fibrous septa develop and lead to micronodular cirrhosis. There is usually minimal inflammation.1
Secondary hemochromatosis is most commonly associated with numerous blood transfusions, usually because the patient has ineffective erythropoeisis (ie. thalassemia, sickel cell disease, aplastic anemia). Interestingly, in secondary hemochromatosis, the iron deposition is seen initially in the Kupffer cells (rather than periportal hepatocytes) and will involve the hepatocytes in later stages.2 However, in many instances, you will not be able to distinguish primary from secondary hemochromatosis and one must rely in clinical history and/or perform genetic testing.
Hereditary hematochromatosis shows a male predilection, being approximately five times more common in men. Furthermore, symptoms appear earlier in men than women due to the protective effects of menstruation in women. The average age of presentation is between 40-60.
Clinical presentation includes abdominal pain, skin pigmentation, diabetes due to destruction of pancreatic islet cells ("bronze diabetes"), cardiomyopathy, arthritis and/or hypogonadism. Lab studies reveal high serum iron and ferritin. A liver biospy would demonstrate increased iron deposition. If the disease is diagnosed early via screening, damage to tissues can be prevented by regular phlebotomy.1
• Liver : Secondary iron overload
1 Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005: 908, 910.
2 Iacobuzio-Donahue CA, Montgomery EA. Gastrointestinal and Liver Pathology: Foundations in Diagnostic Pathology. Philadelphia, PA: Elsevier; 2005: