
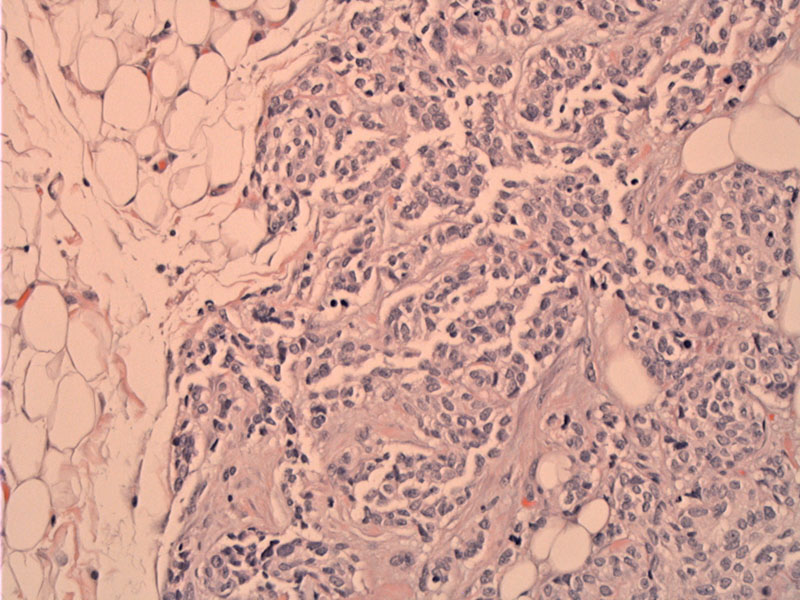
Nests of tumor cells are seen infiltrating into peritoneal adipose tissue. The tumor cells are uniform with scant cytoplasm, indistinct cell borders with hyperchromatic nuclei.
Islands of neoplastic cells arranged in nests and trabeculae are separated by desmoplastic stroma.
Irregular aggregates of primitive appearing cells have hyperchromatic nuclei, indistinct nucleoli and scant cytoplasm.
Yet another view of islands of neoplastic small round blue cells delimited by a dense desmoplastic stroma.
Mitotic figures are usually easily identified. These neoplastic cells have assumed a polygonal shape with indistinct cell borders. Occasionally, tumor cells may assume a rhabdoid appearance i.e. eosinophilic cytoplasmic inclusions and an eccentric nucleus (Rosai, Fletcher).
EMA, an epithelial marker, is intensely positive. DSRCT demonstrates epithelial, muscle and neural lines of differentiation.
CD 57 (Leu-7), a neural marker, is also frequently positive.
Desmoplastic small round cell tumor (DSRCT) arises in the peritoneum, although rare cases in the pleura and nonserosal locations have been described. DSRCT is a member of the family of small round cell tumors which include Ewing sarcoma, primitive neuroectodermal tumor (PNET), rhabdomyosarcoma, neuroblastoma, lymphoma, synovial sarcoma and blastemic Wilms tumor.
The characteristic molecular alteration associated with DSCRCT is the fusion of the Ewing sarcoma gene (EWS) on chromosome 22 to the Wilms tumor gene (WT1) on chromosome 11, leading to the reciprocal translocation t(11;22)(p13;q12), resulting in the EWS-WT1 fusion gene (Kumar, Fletcher).
Parenthetically, a translocation involving the EWS gene is also seen in Ewing sarcoma family of tumors, which includes Ewing sarcoma, extraosseous EWS, primitive neuroectodermal tumor (PNET), neuroepithelioma and Askin tumor (PNET of the chest wall). The translocation partner, however, is different. In EWSFT, the EWS tranlocation partners are 11q24 (FLI1 gene) or 21q22 (ERG gene) and these two translocations are seen in over 85% of EWSFT tumors. In contrast, the translocation partner of DSRCT is the WT1 gene located on 11p13 (Letson).
Grossly, DSRCT can be plaque-like or nodular. The outer surface is usually smooth and bosselated with a firm, grey-white cut surface. Necrosis and myxoid change may be present. These tumors can reach enormous proportions (up to 40 cm) and peritoneal studding may be present. Direct invasion into organs are occasionally seen (Fletcher).
The tumor is thought to arise from a mesothelial progenitor gene capability of multilineage differentiations, thus, DSRCTs can express epithelial (EMA, LMWCK), neuronal (NSE, synaptophysin), and muscle (desmin, vimentin) markers. WT1 staining (targeted toward the C-terminus of the WT1 protein) is also positive (Rosai, Fletcher). The best confirmatory test is via PCR to demonstrate the EWS-WT1 translocation t(11;22)(p13;q12).
Interestingly, antibodies against MIC2 (CD99) antigen can be positive, but the staining pattern is usually cytoplasmic, rather than the membranous staining seen Ewing sarcoma/peripheral neuroectodermal tumor. A hallmark of EWS and PNET is the expression of CD99/MIC2, a cell surface glycoprotein (Letson).
More common in males with M:F ratio of 4:1. Typically affects adolescents and young adults. Presentation includes abdominal pain and sometimes a hard palpable mass and ascites.
Surgical resection combined with chemotherapy and radiation may prolong survival in some but overall prognosis remains poor.
Very poor prognosis. Most patients die within 2 years.
→The cytogenetic lesion is EWS-WT1 t(11;22).
→Thought to arise from a mesothelial progenitor cell with capacity for multilineage differentiation.
→Predilection for the serosal membranes of young adults.
→Poor prognosis and frequent metastases to lymph nodes, lungs and bones.
Chang F. Desmoplastic small round cell tumors: cytologic, histologic, and immunohistochemical features. Arch Pathol Lab Med. 2006 May;130(5):728-32.
Fletcher CDM, ed. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Elsevier; 2007: 889-890.
Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005: 873.
Letson GD, et al. Genetic and Molecular Abnormalities in Tumors of the Bone and Soft Tissues: eMedicine. Last updated on 5/1/2001. Available at: medscape.com/viewarticle/409051
Rosai, J. Rosai and Ackerman's Surgical Pathology. 9th Ed. Philadelphia, PA: Elsevier; 2004: 2382-4.

){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}