
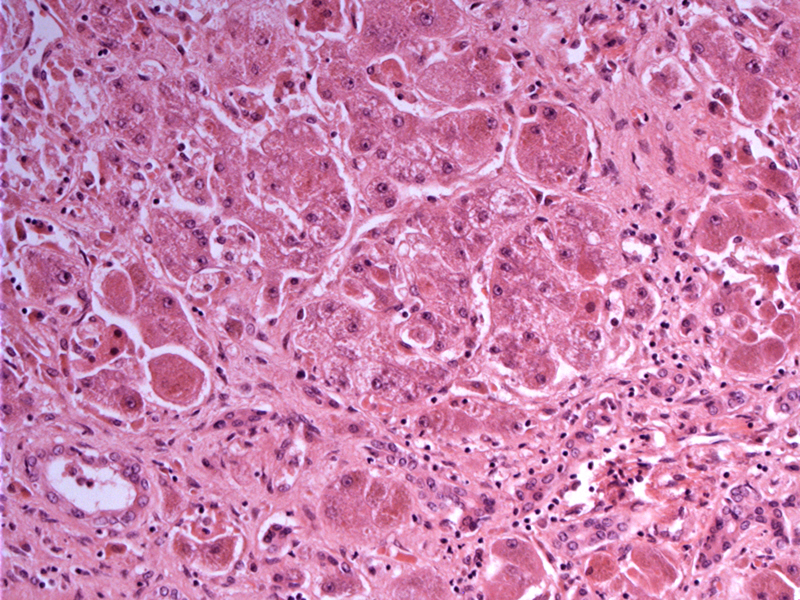
There is anisonucleosis (variation in size of nuclei) of hepatocytes. Intra-sinusoidal collagen is noticeable as well as portal fibrosis. Note that the histologic findings in Wilsons disease are nonspecific, thus, a high degree of suspicion is mandated.
){kind=link}
There is increased lipochrome pigment (lipofuscin) in the hepatocytes, -- note that copper is not visible in regular H&E processing and requires special stains.
){kind=link}
Portal fibrosis is evident and will eventuate in bridging fibrosis and cirrhosis over time.
){kind=link}
This other case shows proliferationg bile ducts and a mild mixed inflammatory portal infiltrate.
){kind=link}
Some steatosis is also evident. There is fibrosis of the portal tracts.
){kind=link}
Wilson disease is an inherited disorder of copper metabolism characterized by excessive copper deposition in the brain, liver and other organs. This autosomal recessive disorder is due to a mutation in the ATPase (transport protein of copper located on chromosome 13qj) that secretes copper into the bile for elimination. Dysfunction of this protein leads to excessive copper accumulation and deposition.
The pathologist must have a high index of suspicion as gross and histologic findings may be nonspecific. Especially in a patient younger than 40 with unexplained chronic liver disease, Wilson's disease should be on the differential (Shah). Histologically, the constellation of findings include micro- or macrovesicular steatosis, an increased number of lymphocytes in the portal tracts, ballooning change, glycogentated nuclei and anisonucleosis (variations in the size of the nuclei).
Copper deposition cannot be seen on regular H&E and require special stains that highlight copper deposition such as rhodanine, rubeanic acid or Shikata stains (Iacobuzio). Later in the course of the disease, cirrhosis and cholestasis may be evident.
Affects both genders equally and the heterozygote carrier rate is 1:100, with the majority of affected persons being compound heterozygotes (Iacobuzio, Shah).
Presentation with hepatic disease such as elevated LFTs and insidious cirrhosis is most commonly seen in children and adolescents (peak age of 10-13). In rare instances, the inital presentation may be fulminant hepatic failure due to sudden release of copious amounts of copper from the liver (Iacobuzio).
Neurological and neuropsychiatric signs usually present a decade later, with concomitant liver findings. One may also see Kayser-Fleischer rings, granular brownish discoloration along the periphery of the iris due to deposition of copper. Laboratory findings that support the diagnosis include ceruloplasmin concentration of less than 0·2 g/L (normal laboratory range 0·2 to 0·5 g/L) as well as elevated urinary copper concentrations.
Lifelong chelation with penicillamine is indicated. Liver transplantation is indicated for end-stage liver disease or fulminant liver failure.
Histological findings of Wilson's disease are non-specific, so a diagnosis requires a high degree of suspicion.
Wilson's disease is an inherited autosome-recessive disorder of copper metabolism.
Deposition of excessive copper in the brain and liver leads to neuropsychiatric and hepatic manifestations.
The mutation is located on chromosome 13q and codes of a copper-transporting ATPase.
Iacobuzio-Donahue CA, Montgomery EA. Gastrointestinal and Liver Pathology: Foundations in Diagnostic Pathology. Philadelphia, PA: Elsevier; 2005: 505-10.
Shah R, Piper MH. Wilson Disease: eMedicine. Last updated August 25 2009. Available at:
http://emedicine.medscape.com/article/183456-overview Accessed on Sept 5 2010.