
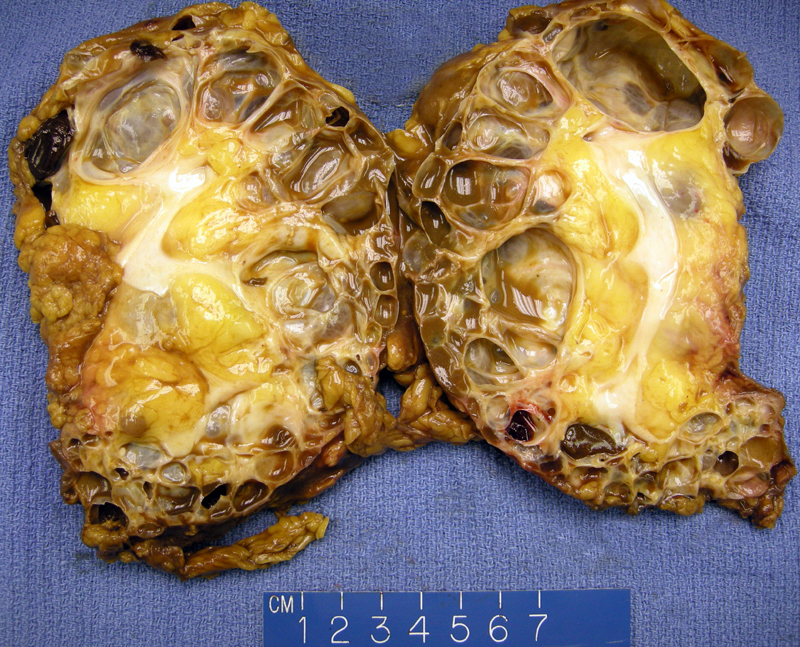
ADPKD is characterized by massively enlarged kidneys (2000 to 4000 grams) with numerous cysts in the cortex and meulla. This example shows a 450 gm nephrectomy with numerous thin wall cysts; the cortex and medulla completely obliterated by cysts. The destruction of renal parenchyma leads to chronic renal failure.
){kind=link}
There is marked parenchymal atrophy with a rare glomerulus on the left. The intervening parenchyma may be normal early in disease and eventually becomes fibrotic, exhibiting tubular atrophy, glomerulosclerosis and vascular sclerosis (Zhou).
){kind=link}
Variably-sized cysts are seen, some containing eosinophilic proteinaceous fluid. The cysts are lined by attenuated cuboidal epithelium. Occasionally, papillary epithelial formations and polyps project into the cystic lumen. Protein, RBCs or calcifications may be seen inside the cysts. A cyst lining is seen with proteinacous fluid. There is interstitial fibrosis and atrophic tubules in the intervening parenchyma.
){kind=link}
Some cyst walls are undermined by xanthomatous inflammation.
){kind=link}
Here is an area of larger cysts with attenuated epithelial lining.
){kind=link}
Cuboidal epithelium is focally present with clear cytoplasm.
){kind=link}
Small infoldings from collections of histiocytes is seen, creating a polypoid projection into the cystic lumen.
){kind=link}
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is common, affecting 1 in 400-1000 live births and accounts for up to 10% of chronic renal failure patients requiring dialysis or renal transplantation (Kumar, Zhou). ADPKD is caused by mutations in PKD1 on chromosome 16p13.3 (85-90% of cases) and PKD2 (10-15%) on chromosome 4q21. These polycystin genes are membrane-associated glycoproteins involved in cell-cell and cell-matrix interactions. Mutations in PKD1 lead to more severe disease with more rapid disease progression. PKD1 patients experience renal failure by age 53 compared to age 69 in PKD2 patients (Kumar).
Age at presentation varies, but symptoms usually appear by 3rd or 4th decade; there is 100% penetrance by age 80. In 25% of cases, ADPKD develops from a new de novo mutation and a family history is lacking.
Symptoms include flank pain due to enlarged kidneys or acute pain from hemorrhage into one of the cysts. Hypertension and hematuria often develops, and urinary tract infections develops in 50 to 70% of patients. Perinephric abscesses is a serious complication that has a 60% mortality rate. Renal cell carcinoma develops in 1-5%.
Extrarenal involvement is common in ADPKD. 40% of patients have polycystic liver disease, in which cysts are derived from the biliary epithelium. Cysts may also develop in the pancreas and spleen as well. Intracranial berry aneurysms arise in the Circle of Willis and may lead to death from subarachnoid hemorrhage in 4 to 10% of patients (Kumar). Valvular disease (e.g. floppy mitral valve) may also be seen.
Dialysis and renal tranplantation prolongs life. Extrarenal complications must be monitored (ie. infection of kidneys, hypertensive cardiovascular disease, berry aneurysms), but there is no cure.
• Kidney : Autosomal Recessive Polycystic Kidney Disease
Cheng L, Bostwick DG, eds. Essentials of Anatomic Pathology. 2nd Ed. Totowa, NJ: Humana Press; 2006: 1143-4.
Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005: 962-3.
Zhou M, Magi-Galluzzi, C. Genitourinary Pathology: Foundations in Diagnostic Pathology. Philadelphia, PA: Elvesier; 2006: 229-232.