
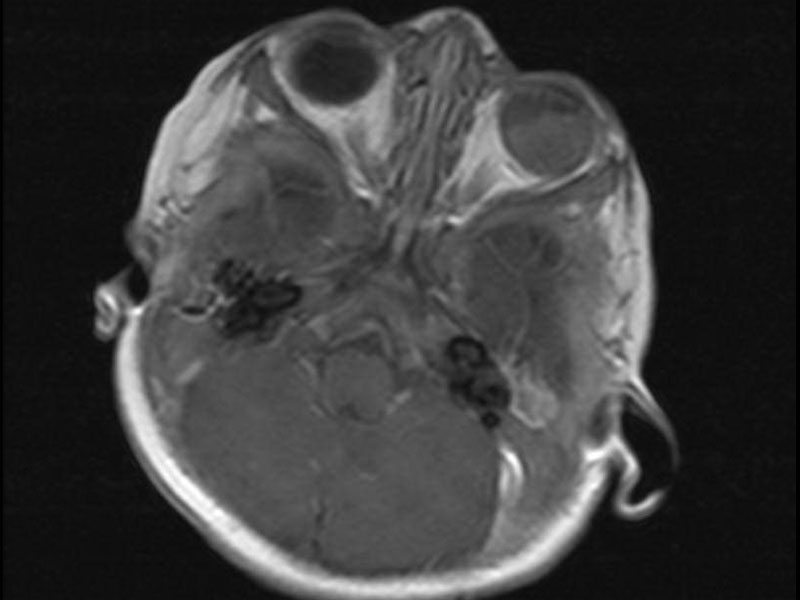
MRI shows an abnormal left globe with 70% involvement by an irregular mass principally involving the posterior lateral aspect.
){kind=link}
Case 1 demonstrates Flexner-Wintersteiner rosettes, which have a central lumen.
){kind=link}
Punctate tumor necrosis is seen within cellular areas -- this alternation of cellular areas and necrotic islands is characteristic of retinoblastoma.
){kind=link}
Case 2 demonstrates a diffuse proliferation of small round blue cells with overlying retinal epithelium. A hint of rosette formation can be seen.
){kind=link}
Case 3 is yet another view of a proliferation of neoplastic retinoblastoma cells. The outer nuclear layer and inner nuclear layer of the retina can be seen as two ribbon-like bands of darkly staining cells.
){kind=link}
Zones of necrosis and calcification are common findings in retinoblastoma.
){kind=link}
Retinoblastoma, the most common ocular malignancy in children, is the prototypical example for the "two-hit" hypothesis of oncogenesis. Familial cases of retinoblastoma occur when there is a germline mutation of the RB gene (i.e. the first "hit"). Subsequent somatic mutation of the second allele (i.e. the second "hit") results in a cell having two defective (or absent) copies of the Rb gene, a tumor suppressor gene -- thus, allowing uncontrolled cellular proliferation (Kumar).
RB, one of the "small blue round cell tumors" of childhood, is composed of sheets of undifferentiated small dark cells. At low power, there should be alternating zones of cellularity and necrosis with scattered calcifications. It is important to mention calcifications (if found) in the final pathology report because the clinicians often use the presence of intraocular calcium deposits to diagnose the tumor (Fletcher). RB is very vascular and the neoplastic cells tend to cluster around vessels and can lead to deposition of nuclear material around vessel walls, reminiscent of the Azzopardi effect seen in small cell carcinoma of the lung.
In well-differentiated tumors, several types of rosettes may be seen: (1) Flexner-Wintersteiner rosettes (most common in RB) -- composed of a single layer of tumor cells arranged around a central lumen); (2) fleurettes -- a single layer of cells with tapered cytoplasmic processes protruding into the center of the lumen; (3) Homer Wright rosettes -- single layer of cells around a central core of fibers with no true lumen.
Some areas should still form characteristic Flexner-Wiintersteiner or Homer-Wright rosettes.
Approximately 250-500 new cases of retinoblastoma occur in the United States each year. The average age at diagnosis is 18 months with 90% of cases diagnosed prior to age 5. Clinical presentations includes leukocoria (white pupillary reflex) and strabismus (Isidro).
Patients with inherited (familial retinoblastoma) are at increased risk of developing other tumors such as osteosarcoma. Patients with these germline mutations are more likely to have bilateral tumors and "trilateral retinoblastoma" which is a pinealoblastoma.
Conservative surgical excision and adjuvant chemotherpay are employed, with a view toward preserving eyesight in these young patients.
Invasion in to the optic nerve, extraocular extension and possibly choroidal invasion predicts a more aggressive tumor (Kumar, Fletcher). The degree of differentiation does not appear to affect prognosis.
Fletcher CDM, ed. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Elsevier; 2007: 1802-4.
Isidro MA, et al. Retinoblastoma: eMedicine. Last updated Sept 21 2010. Available at: emedicine.medscape.com/article/1222849-overview
Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005: 299,